DISTURBANCES in mineral and bone metabolism are common in patients with chronic kidney disease (CKD). Table 2 lists the major features of these abnormalities.4-15 The processes causing disordered mineral metabolism and bone disease have their onset in the early stages of CKD, continue throughout the course of progressive loss of kidney function, and may be influenced beneficially or adversely by various therapeutic approaches used.
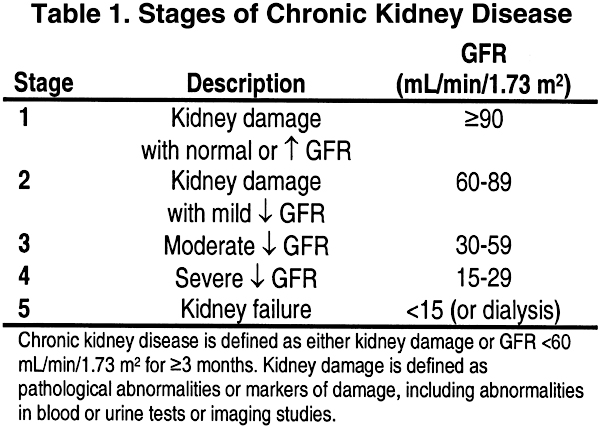
In this section and those that follow, the stage of CKD is defined according to the Clinical Practice Guidelines for Chronic Kidney Disease: Evaluation, Classification, and Stratification (Table 1).
Hypocalcemia and Secondary Hyperparathyroidism
Patients with CKD almost always develop secondary hyperplasia of the parathyroid glands, resulting in elevated blood levels of parathyroid hormone (PTH). This abnormality is due to the hypocalcemia that develops during the course of kidney disease and/or to a deficiency of 1,25-dihydroxycholecalciferol [l,25(OH)2D3] that may directly affect the function of the parathyroid glands. With progressive loss of kidney function, a decrease in the number of vitamin D receptors (VDR) and calcium-sensing receptors (CaR) in the parathyroid glands occurs, rendering them more resistant to the action of vitamin D and calcium. In addition, the development of hyperphosphatemia directly affects the function and the growth of the parathyroid glands. These events will allow secondary hyperparathyroidism to worsen.
At least 3 hypotheses have been proposed to explain the pathogenesis of the hypocalcemia: (a) phosphate retention, (b) skeletal resistance to the calcemic action of PTH, and (c) altered vitamin D metabolism. The zeal and vigor with which the proponents of these hypotheses have defended these concepts have created the impression that a major controversy exists in the pathogenesis of hypocalcemia and secondary hyperparathyroidism. However, these possibilities are not mutually exclusive but rather interrelated. Together, these factors form a unified and integrated explanation for the hypocalcemia of CKD, and provide a framework for the management of altered mineral and bone metabolism of CKD.
Role of phosphate retention. Several lines of evidence suggest that phosphate retention can provoke secondary hyperparathyroidism. First is a disorder called sneezing disease described in piglets ingesting high-phosphate diets. This disease is characterized by labored respiration and sneezing and is due to deformities of turbinate nasal bones caused by generalized osteitis fibrosa. This disease was reproduced in horses fed high-phosphate, low-calcium diets. The animals developed lameness and a “big head” secondary to swelling of facial bones; both abnormalities were due to osteoclastic bone resorption. Initially, these animals developed hyperphosphatemia and hypocalcemia followed by hypophosphatemia and hypercalcemia. Their parathyroid glands were diffusely hyperplastic. These observations demonstrate that the ingestion of an excessive amount of phosphate is associated with secondary hyperparathyroidism, even in the absence of CKD.
Second, the acute ingestion of inorganic phosphate by normal subjects has been shown to cause a transient rise in the levels of serum phosphorus, a fall in the concentration of ionized calcium, and a significant elevation in the blood levels of PTH even in the presence of normal kidney function.
Third, the development of secondary hyperparathyroidism in dogs with experimentally-induced reduction in kidney function is influenced by the magnitude of dietary phosphate intake; and the secondary hyperparathyroidism was prevented when dietary intake of phosphate was reduced in proportion to the experimentally induced reduction in the glomerular filtration rate (GFR).
It is evident, therefore, that phosphate retention and hyperphosphatemia can provoke secondary hyperparathyroidism in the absence or presence of impaired kidney function. Consequently, because secondary hyperparathyroidism occurs early in the course of CKD, hyperphosphatemia would be expected to develop at an early stage of reduced kidney function. However, the available data indicate that patients with moderate loss of kidney function (Stage 3) are not hyperphosphatemic but are either normophosphatemic or mildly hypophosphatemic. To explain this discrepancy, it was postulated that a transient and possibly undetectable increase in serum phosphorus occurs early in CKD with each decrement in kidney function. Such a transient hyperphosphatemia would directly decrease the blood levels of ionized calcium, which then stimulates the parathyroid glands to release more hormone (PTH). The elevation in the blood levels of PTH would decrease the tubular reabsorption of phosphate in the kidney and increase the excretion of phosphate in the urine, with the return of serum phosphorus and calcium levels to normal, but at the expense of a new steady state characterized by elevated blood levels of PTH.
This postulate implies that the adaptive changes occurring in patients with incipient loss of kidney function, and leading to secondary hyperparathyroidism, are geared to maintain normal phosphate homeostasis. However, ample evidence exists indicating that phosphate homeostasis in CKD can be maintained without secondary hyperparathyroidism. First, in thyroparathyroidectomized dogs with experimentally induced reduction in kidney function in which the serum calcium was maintained at a normal level by vitamin D supplementation, the fraction of filtered phosphate excreted by the kidney increased and the serum concentration of phosphorus remained normal despite the loss of kidney function. Second, rats immunized against tubular basement membrane developed interstitial nephritis and reduced kidney function, and the kidneys lost the ability to generate cyclic AMP in response to PTH. Despite these changes, the fraction of filtered phosphate excreted in the urine increased markedly. Third, studies in rats with reduced kidney function have clearly demonstrated that PTH is not the main regulator of the handling of phosphate by the kidney. Fourth, a very high fractional excretion of phosphate is present in patients with CKD, even after total parathyroidectomy.

If the sequence of events described by the phosphate retention hypothesis does occur, the basal serum levels of phosphorus and calcium should display one of the following combinations: hyperphosphatemia and hypocalcemia, normophosphatemia and normocalcemia, or hypophosphatemia and hypercalcemia. The latter may occur if the adaptive response of the parathyroid glands is exaggerated, as in the case of nutritionally induced secondary hyperparathyroidism in horses. However, available data show that the mean levels of both serum phosphorus and calcium in most patients with moderate loss of kidney function are actually lower than the values in normal subjects. These observations cannot be explained by the phosphate retention theory alone. Thus, other factors must also be operative and contribute to the genesis of the hypocalcemia in the early course of CKD.
These considerations do not necessarily mean that phosphate retention is not an important factor in the pathogenesis of the hypocalcemia and secondary hyperparathyroidism of CKD. Rather, they suggest that phosphate retention in the course of CKD may contribute to the hypocalcemia by mechanisms other than a direct effect of hyperphosphatemia on serum calcium.
It should be mentioned that with more advanced loss of kidney function (Stages 4 and 5) when hyperphosphatemia develops, the elevated blood levels of phosphorus may suppress blood levels of calcium and contribute to the hypocalcemia. In addition, experimental evidence indicates that the very high levels of serum phosphorus may directly affect the function of the parathyroid glands; such high serum phosphorus levels may induce hyperplasia of the parathyroid glands independent of hypocalcemia and/or reduced blood levels of 1,25(OH)2D3. Hyperphosphatemia has a direct effect on post-transcriptional mechanisms that increase PTH synthesis and secretion.
Role of skeletal resistance to the calcemic action of PTH.The calcemic response to the infusion of PTH or to an acute rise in the blood levels of endogenous PTH is markedly blunted in patients with mild to moderate loss of kidney function (creatinine clearance 25 to 85 mL/min/1.73 m2 [0.42 to 1.42 mL/s/1.73 m2]), indicating that this skeletal resistance occurs early in the course of loss of kidney function. This abnormality has also been documented in patients with severe loss of kidney function (creatinine clearance of less than 20 mL/min/1.73 m2 [0.33 mL/s/1.73 m2]) and in those treated with hemodialysis and in many kidney transplant recipients whose kidney function is usually below normal (creatinine clearance 71 ± 6.7 mL/min/1.73 m2 [1.18 ± 0.11 mL/s/1.73 m2]).Skeletal resistance to the calcemic action of PTH occurs in patients with acute kidney failure as well. Hypocalcemia is almost always observed in these patients. The degree of hypocalcemia is moderate to marked (range, 7.5 to 8.0 mg/dL) and lower levels have also been reported. The hypocalcemia occurs early in the course of the oliguric phase of the disease and persists through the diuretic period. This hypocalcemia is observed in patients with low, normal, or elevated serum concentrations of phosphorus, indicating that the hyperphosphatemia of acute kidney failure is not the major determinant of the hypocalcemia. Also, the hypocalcemia cannot be attributed to a failure in the function of the parathyroid glands because the blood levels of PTH are elevated and display an inverse correlation to the concentrations of serum calcium. Further, the infusion of PTH fails to elicit a normal rise in serum calcium. All these derangements are reversed after the return of kidney function to normal.These observations indicate that there is a skeletal resistance to the calcium-mobilizing action of PTH, an abnormality that occurs early in the course of both acute and chronic kidney disease and is not reversed by hemodialysis. This derangement is an important factor contributing to the hypocalcemia in kidney disease and to the pathogenesis of secondary hyperparathyroidism in these patients.
A series of studies in thyroparathyroidectomized dogs with diverse models of acute kidney failure (bilateral ureteral ligation, bilateral nephrectomy, or diversion of both ureters into the jugular veins) has demonstrated that the skeletal resistance to the calcemic action of PTH is partially due to a deficiency of 1,25(OH)2D3 and its complete correction requires adequate amounts of both 1,25(OH)2D3 and 24,25(OH)2D3. Other studies suggest that the skeletal resistance to the calcemic action of PTH is, at least in part, due to downregulation of PTH receptors. Indeed, several studies have shown that the PTH-PTHrP receptors are downregulated in many organs in uremia; these include the kidney, liver, and heart. This downregulation of the PTH-PTHrP receptors is not due to the high blood levels of PTH but rather to the PTH-induced elevation in the basal levels of intracellular concentrations of calcium (cytosolic calcium) in those organs. Indeed, in kidney failure the basal levels of cytosolic calcium is elevated in all organs, and the correction of this abnormality by treatment with calcium channel blockers is associated with reversal of the downregulation of the PTH-PTHrP receptors.
Role of altered vitamin D metabolism. The experimental data cited previously suggest that alterations in vitamin D metabolism and/or a deficiency of 1 or more of the vitamin D metabolites are present in patients with early CKD (Stage 2 and 3) because these patients display skeletal resistance to the calcemic action of PTH. Indeed, disturbances in the functional integrity of the target organs for vitamin D (impaired intestinal absorption of calcium and/or defective mineralization of osteoid) have been found in patients with mild CKD (Stage 2 and 3), indicating that a state of relative or absolute vitamin D deficiency exists in these patients.
The blood levels of l,25(OH)2D3 are usually normal or modestly elevated in patients with moderate CKD (creatinine clearance >50 mL/min/1.73 m2 [0.83 mL/s/1.73 m2]), although low levels have also been noted in both adults and children with these levels of kidney function. Therefore, it appears that absolute deficiency of and/or resistance to vitamin D [normal blood levels of 1,25(OH)2D3] develop early in the course of CKD. As mentioned earlier, the number of VDRs decreases as the loss of kidney function progresses, resulting in resistance to vitamin D action. The blood levels of 1,25(OH)2D3 in Stage 4 of CKD are definitely low and are usually undetectable in the dialysis patients.
It is intriguing that, despite the presence of adequate functioning kidney mass in patients with moderate reduction in kidney function (Stage 2), the production of l,25(OH)2D3 does not increase adequately to meet the needs of the target organs for vitamin D. Because the regulation of the kidney lα-hydroxylase, the enzyme responsible for 1,25(OH)2D3 production, is influenced by alterations in phosphate homeostasis, it is possible that phosphate retention, which may develop with declining kidney function, plays a role in the disturbances in 1,25(OH)2D3 production. Indeed, dietary phosphate restriction in proportion to the reduction in GFR in adults with Stage 2 CKD has been associated with a significant increase in the blood levels of l,25(OH)2D3 and with biological evidence for the normalization of the target organ response to vitamin D.
The mechanisms through which dietary phosphate restriction in patients with Stage 2 CKD is associated with increased production of l,25(OH)2D3 are not evident. This effect does not seem to be mediated by changes in the serum levels of phosphorus because no significant changes in this parameter were found in adults. The effect of dietary phosphate on kidney production of l,25(OH)2D3 could be mediated through changes in transcellular flux of phosphate and/or in the concentration of inorganic phosphorus in kidney cortical cells. Indeed, studies in rats have shown that the level of inorganic phosphorus in the kidney cell is reduced during the feeding of a phosphate-restricted diet.
Interaction between 1,25(OH)2D3 and parathyroid glands. Available data indicate that l,25(OH)2D3 may have a direct effect on the parathyroid glands. First, exposure to 1,25(OH)2D3 both in vivo and in vitro may directly suppress the activity of the parathyroid glands. Second, l,25(OH)2D3 renders the parathyroid glands more susceptible to the suppressive action of calcium. Such an effect of l,25(OH)2D3 may correct the abnormal shift in set point for calcium of the parathyroid glands in patients with CKD. Third, 1,25(OH)2D3 decreases prepro-PTH messenger RNA in a dose-dependent manner. Thus, it is possible that deficiency of 1,25(OH)2D3 may initiate secondary hyperparathyroidism even in the absence of overt hypocalcemia; this has been demonstrated in dogs with reduced kidney function.
Regulation of the parathyroid hormone gene by vitamin D, calcium, and phosphorus. Secondary hyperparathyroidism in CKD is due to increased synthesis and secretion of PTH secondary to an increase in PTH gene expression and parathyroid cell proliferation. 1,25(OH)2D3 acts directly on the PTH gene, causing a decrease in its transcription and hence in the synthesis of PTH. Hypocalcemia increases and hypophosphatemia decreases PTH gene expression by an effect on the stability of the PTH mRNA. Thus, there is an increase in the stability PTH mRNA associated with hypocalcemia, resulting in increased synthesis of the PTH protein. In contrast, the stability of PTH mRNA is decreased during hypophosphatemia, leading to increased degradation of the PTH mRNA and hence decreased production of PTH. Thus, the effects of hypercalcemia and hypophosphatemia on PTH synthesis is post-transcriptional.
Integration of the Various Pathogenetic Factors in the Genesis of Secondary Hyperparathyroidism (Fig 1)
The clinical and experimental evidence considered thus far allow an integrated formulation for the mechanisms of secondary hyperparathyroidism in CKD. It appears that phosphate retention, which may develop with loss of kidney function, interferes with the ability of patients with CKD to augment the production of 1,25(OH)2D3 by the kidneys to meet the increased need for this metabolite. Thus, a state of absolute or relative vitamin D deficiency develops, leading to defective intestinal absorption of calcium and impaired calcemic response to PTH. These 2 abnormalities produce hypocalcemia which in turn causes secondary hyperparathyroidism. Although this formulation still assigns an important role to phosphate retention in the genesis of secondary hyperparathyroidism in CKD, the pathway through which such phosphate retention mediates its effect is different from that originally proposed. The original theory maintained that phosphate retention in the early course of CKD is associated with a rise in levels of serum phosphorus and a consequent fall in the levels of serum ionized calcium, which in turn stimulates the parathyroid gland activity. However, it must be emphasized that if marked hyperphosphatemia does develop in a patient with CKD, it could directly lower the level of serum calcium and contribute to the severity of the hypocalcemia and the secondary hyperparathyroidism. In addition, hyperphosphatemia per se may stimulate parathyroid hormone synthesis by a post-transcriptional effect on PTH gene expression. An Na-P cotransporter is present in the parathyroid gland, and this transporter may play a role in the process that allows the parathyroid gland to sense the level of extracellular phosphorus.
An additional pathway through which an absolute or relative deficiency of 1,25(OH)2D3, independent of hypocalcemia, may mediate secondary hyperparathyroidism is related to its direct effect on the parathyroid glands, as discussed earlier.
This integrated formulation for the pathogenesis of secondary hyperparathyroidism has important clinical implications. It is consistent with the hypothesis that dietary phosphate restriction in proportion to the fall in GFR in patients with CKD is adequate to reverse and correct secondary hyperparathyroidism and other abnormalities in mineral metabolism. However, achieving the proper and adequate dietary phosphate restriction and successful patient compliance with the dietary regimen may prove difficult. Because the available data indicate that dietary phosphate restriction exerts its effect through the increased production of 1,25(OH)2D3 and because this vitamin D metabolite also exerts a direct effect on the parathyroid glands, an alternative therapeutic approach would be supplementation of 1,25(OH)2D3. Indeed, treatment of patients with Stage 3 CKD with 1,25(OH)2D3 for 12 months was associated with improvement or normalization of the disturbances in mineral metabolism, including secondary hyperparathyroidism and bone disease.
Structure and Function of the Parathyroid Glands
Hyperplasia of the parathyroid glands is almost always present in patients with CKD, but the increase in volume and mass of the glands varies among patients and among the 4 glands in the same patient. The size of the glands may reach 10 to 50 times normal. Occasionally, the parathyroid glands may be of normal size in patients with CKD. Histologically, the glands show chief cell hyperplasia with or without oxyphil cell hyperplasia. The usual cell is the vacuolated or chronically stimulated chief cell, 6 to 8 μm wide, with a sharply defined plasma membrane. Nodular or adenomatous-like masses may be found within the hyperplastic glands. These nodules are well circumscribed and surrounded by a fibrous capsule. The cells in the nodular hyperplasia have less VDR and calcium-sensing receptor (CaR) density and a higher proliferative potential than the cells of diffuse hyperplasia. The change in the structure of the parathyroid glands begins as polyclonal diffuse hyperplasia. The cells with the lower density of VDR and CaR start to proliferate monoclonally (early nodularity in diffuse hyperplasia) and form nodules. Several monoclonal nodules of different size may develop resulting in multinodular hyperplasia. Alternatively, the cells of 1 of the nodules may proliferate faster and more vigorously giving rise to a very large nodule that almost occupies the entire gland (single nodular gland). Molecular changes are implicated in the tumorigenesis of the parathyroid gland in CKD. However, the exact abnormalities underlying the monoclonal cell proliferation and the biochemical and molecular processes responsible for the differences in the proliferative potentials of these nodules are, as yet, not elucidated.
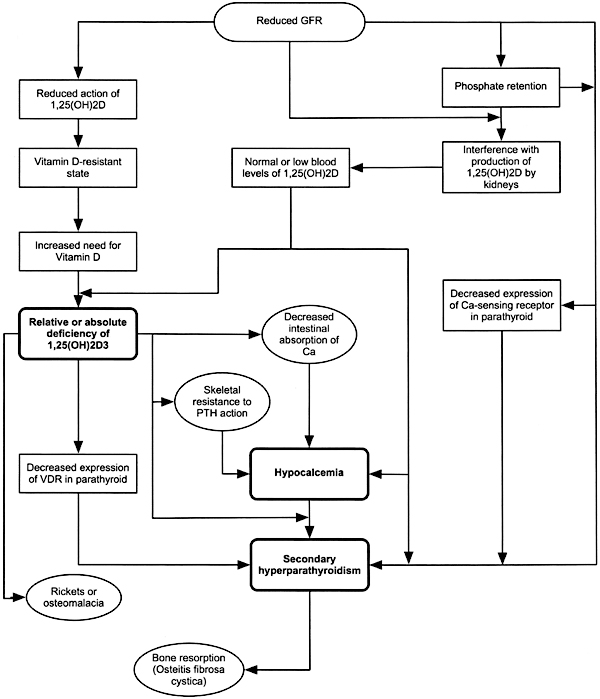
Fig 1. Pathogenesis of abnormalities in mineral metabolism and bone disease in CKD.
Hypocalcemia, relative or absolute deficiency of 1,25(OH)2D3, and phosphate retention or hyperphosphatemia are the most important factors responsible for the hyperplasia of the parathyroid glands. Because hypocalcemia and relative or absolute deficiency of 1,25(OH)2D3 (vitamin D resistance) may develop early in the course of CKD, hyperactivity of the parathyroid glands is also encountered in the early stages of kidney disease. Indeed, elevated blood levels of PTH may be noted when the GFR falls below 60 mL/min/1.73 m2.
The appearance of spontaneous and persistent hypercalcemia in some uremic patients (Stage 4 and 5 CKD) has led to the suggestion that the parathyroid glands in these patients may ultimately become autonomous. However, after calcium infusion, the blood levels of PTH of these patients invariably fall, but not to normal levels. Thus, the parathyroid glands in these patients are suppressible at higher levels of serum calcium. The appearance of spontaneous hypercalcemia and the failure of the blood levels of PTH to fall to normal values after calcium infusion in uremic patients (Stage 4 and 5 CKD) are most likely due to the large mass of the parathyroid glands in these individuals. Malregulation of PTH release at the cellular level may also be present. Indeed, in vitro studies of dispersed cells from the parathyroid glands of such patients show that a higher concentration of calcium is required to achieve a suppression of PTH secretion. This has been interpreted to indicate a shift in the set point for calcium; this abnormality is at least in part due to deficiency in 1,25(OH)2D3. True adenomas may develop and function autonomously in certain cases of secondary hyperparathyroidism, but such cases are not common. The use of the term tertiary hyperparathyroidism should be limited to those cases in which it is documented that a true adenoma has developed in a previously hyperplastic gland.
Multiple factors control the release of PTH from the glands, and they do so by inducing changes in the cellular function of the parathyroid glands. Calcium is the most important regulator of PTH secretion, and its effect is mediated by changes in intracellular concentration of calcium. An increase in the latter influences PTH secretion through several cellular mechanisms such as inhibition of cAMP accumulation or its action, and/or the stimulation of intracellular degradation of preformed PTH.
The CaR is located in the membrane of the cells of the parathyroid glands. This receptor protein plays an important role in the ability of parathyroid glands to recognize changes in the concentration of calcium ion in the blood and as such, CaR mediates the effect of calcium on the secretion of PTH from the parathyroid glands. In the course of CKD, there is a reduction in the density of CaR in the parathyroid gland cells. The levels of serum calcium and l,25(OH)2D3 as well as dietary phosphate do not appear to regulate the synthesis of CaR.
The relationship between the serum levels of calcium and the parathyroid gland in the modulation of PTH secretion is altered in CKD patients. In normal subjects, this relationship is sigmoidal over a narrow range of calcium concentration, but in patients with CKD, higher levels of serum calcium are needed to suppress the secretion of PTH compared to normal subjects. Also, in CKD patients, the susceptibility of parathyroid adenyl cyclase to the inhibitory effect of calcium is reduced. Such an effect would impair the ability of calcium to inhibit PTH secretion. These abnormalities in calcium and PTH secretion could be evaluated by the changes in set-point for calcium. The latter is defined as the calcium concentration that produces half the maximal inhibition of PTH and that is the midpoint between the maximal and minimal PTH secretions. Indeed, alterations in set-point for calcium with a shift to right (eg, 50% inhibition of PTH secretion occurs at higher calcium concentration) were observed in parathyroid glands of patients with primary or secondary hyperparathyroidism. Administration of 1,25(OH)2D3 to dialysis patients was associated with suppression of PTH secretion and with a shift of the set-point to the left, supporting the hypothesis that deficiency of this vitamin D metabolite plays an important role in the genesis of secondary hyperparathyroidism in CKD.
Thus, the available evidence suggests that in patients with CKD, the structural changes in the parathyroid glands (increase in their mass due to diffuse and nodular hyperplasia) and its functional abnormality (shift in set point of calcium to the right) are responsible to the increase production and release of PTH. Because the changes in the structure and function of the parathyroid glands occur early in the course of CKD, the blood levels of PTH are elevated when the GFR falls below 60 mL/min/1.73 m2.

After its secretion from the parathyroid gland, intact PTH is cleaved by the liver into an N- and a C-terminal fragment. The half-life of both the intact hormone and its N-terminal fragment is short (about 5 minutes), whereas that of the C-terminal fragment is much longer. Stages 4 and 5 of CKD are associated with alterations in PTH metabolism. Both the hepatic removal of the intact hormone and the kidney clearance of the C-terminal fragment are impaired. Thus, the elevated blood levels of PTH in CKD are due to both increased secretion and impaired degradation. The major component of the elevated blood levels of the immunoreactive PTH in these patients is the C-terminal fragments and particularly the midmolecule or midregion of C-terminal fragments.
Hyperplasia of the parathyroid glands in patients with CKD is not easily reversed, even after the correction of its causes. Some investigators found that parathyroid gland hyperplasia regresses in all patients in whom PTH secretion was successfully suppressed. The mechanisms underlying this regression are not well understood. Apoptosis has been proposed, and certain in vitro studies indicate that very high concentrations of 1,25(OH)2D3 induce apoptosis of parathyroid gland cells. Such an effect may occur in vivo as well. This phenomenon has been utilized to achieve medical parathyroidectomy by injecting 1,25(OH)2D3 directly into the hyperplastic parathyroid glands. In some patients, spontaneous hemorrhage in the hyperplastic glands occurs and may be responsible for the regression of the hyperplastic glands in occasional cases.
Hyperphosphatemia
Although phosphate retention occurs early in the course of CKD (Stage 2), hyperphosphatemia becomes evident in patients with marked loss of kidney function (Stage 4). Several factors may affect the level of serum phosphorus in patients with CKD (Table 3).
As mentioned earlier, elevation in serum levels of phosphorus occurs when GFR falls below 30 mL/min/1.73 m2 and the severity of hyperphosphatemia becomes greater with further declines in GFR. The dietary intake of phosphate and the fraction of the ingested phosphate absorbed by the intestine have an important effect on the serum levels of phosphorus in patients with CKD. These patients have only mild impairment in intestinal absorption of phosphate, but their kidneys are unable to adequately handle phosphate loads. Thus, an increase in phosphate intake can cause a marked rise in serum phosphorus levels when GFR falls below 30 mL/min/1.73 m2.
Intestinal absorption of phosphate is enhanced by 1,25(OH)2D3, and its administration to patients in Stages 4 and 5 of CKD may produce or worsen hyperphosphatemia. In patients who have substantial osteomalacia, the levels of serum phosphorus may remain unchanged or even fall during therapy with 1,25(OH)2D3. This is due to the deposition of calcium and phosphorus into bone as 1,25(OH)2D3 improves mineralization of osteoid and heals osteomalacia.
Phosphate-binding compounds render dietary phosphate and phosphate contained in swallowed saliva and intestinal secretions unabsorbable. Thus, patients receiving these compounds may have normal levels of serum phosphorus or develop modest hypophosphatemia. It should be emphasized that these compounds are most effective when dietary intake of phosphate is below 1.0 g/day. With higher phosphate intake (more than 2.0 g/day), their effectiveness is reduced and hyperphosphatemia may persist despite their use.
An important factor determining the level of serum phosphorus in Stage 4 and 5 CKD is the degree of hypersecretion of PTH and the response of the skeleton to the high levels of this hormone. Normally, PTH decreases the tubular reabsorption of phosphate by the kidney, increases urinary phosphate excretion, and consequently maintains serum phosphorus levels. This effect becomes progressively limited as loss of kidney function advances (GFR <20 mL/min/1.73 m2). In such patients, the severely damaged kidneys cannot respond to further increments in PTH with additional augmentation in phosphate excretion. The enhanced bone resorption, which is induced by the high levels of PTH, liberates calcium and phosphorus from the skeleton into the extracellular fluid. This phosphorus cannot be excreted by the kidney and hence serum phosphorus concentration rises. The same phenomenon occurs in dialysis patients. Several clinical observations support this view. First, the levels of serum calcium and phosphorus are higher in patients with advanced kidney failure (Stage 5) and severe secondary hyperparathyroidism than in other patients with comparable kidney failure but without severe hyperparathyroidism. Second, following total or subtotal parathyroidectomy in patients with kidney failure and severe secondary hyperparathyroidism, the serum concentrations of calcium and phosphorus fall (Fig 2). Third, when patients with chronic kidney disease and overt secondary hyperparathyroidism are treated with hemodialysis, the serum phosphorus levels not only may remain above normal but may rebound rapidly after dialysis to predialysis levels.
A shift in the balance between protein synthesis and breakdown toward catabolism, as occurs with infection, trauma, starvation, and the administration of glucocorticoids or tetracycline, can cause an increase in serum phosphorus concentration. The parenteral administration of solutions containing large quantities of glucose and amino acids to such patients cause an abrupt reduction in serum phosphorus levels. Also, the concentration of serum phosphorus may fall during refeeding after a period of calorie or protein malnutrition.
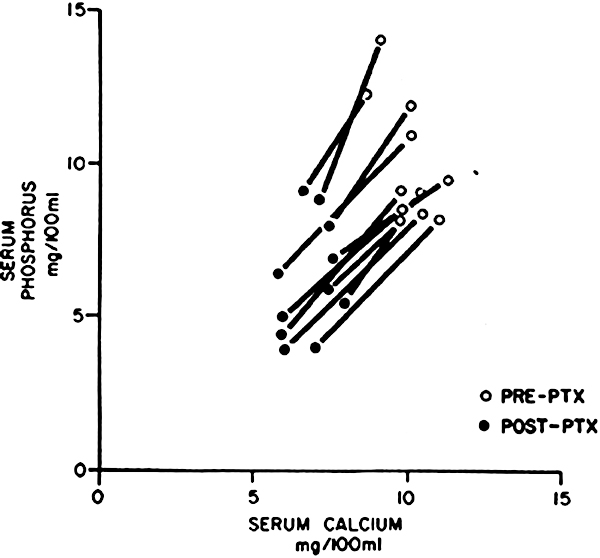
Fig 2. Changes in total serum calcium and inorganic phosphorus observed in 11 uremic patients before and after subtotal parathyroidectomy. Reproduced with permission.16
The use of calcium compounds in patients with Stage 4 and 5 CKD results in the reduction in the serum levels of phosphorus due to the ability of these compounds to bind phosphate in the intestine. In addition, these calcium compounds cause a rise in serum calcium levels which would inhibit the parathyroid gland and results in a fall in blood PTH levels. This would be followed by a reduction in serum levels of serum phosphorus as discussed above.
Altered Vitamin D Metabolism
Patients with Stage 2 and 3 CKD may have a vitamin D-resistant state and/or a relative vitamin D-deficient state. As kidney function deteriorates further, an absolute vitamin D-deficient state develops, with the blood levels of 1,25(OH)2D3 being reduced when GFR falls below 50 mL/min/1.73 m2 (Stage 3) in children and below 30 mL/min/1.73 m2 (Stage 4) in adults. In anephric patients and in those treated with dialysis, the blood levels of 1,25(OH)2D3 are usually undetectable. In advanced CKD (Stages 4 and 5), the number of VDRs is reduced, leading to vitamin D resistance. Thus, in such patients, there is vitamin D deficiency and vitamin D resistance as well.

The blood levels of 25-hydroxyvitamin D [25(OH)D3] in patients with CKD may be low. Low levels of 25(OH)D3 may be encountered in patients who have nephrotic-range proteinuria due to loss of 25(OH)D3 in urine, those who are treated with peritoneal dialysis due to loss of 25(OH)D3in peritoneal fluid, or those who have nutritional vitamin D deficiency.
The biological consequences of vitamin D deficiency are multiple and are manifested by disturbances in the function of its target organs: parathyroid glands, bone, intestine, and skeletal muscle (Table 4). Because other organs such as testes, myocardium, and pancreas have receptors for 1,25(OH)2D3, it is possible that a deficiency of this vitamin D metabolite plays a role in the dysfunction of these organs in kidney failure.
The factors responsible for the decrease in the number of VDRs in kidney failure are not fully elucidated, but may include (a) reduced levels of 1,25(OH)2D3 because this metabolite affects the production of VDRs (low levels of 1,25(OH)2D3, downregulates the mRNA of VDR); (b) hyperparathyroidism of CKD (high levels of PTH interfere with the 1,25(OH)2D3-induced upregulation of VDR); and (c) uremic toxins, which may decrease the stability of the mRNA of VDRs, resulting in reduced expression of VDR protein.
The action of vitamin D is mediated by its binding to its cytosolic receptor, VDR. The DNA binding site for VDR is a nuclear receptor that contains 2 “zinc fingers” that mediate the binding of VDR to a regulatory promoter in regions of DNA upstream of the vitamin D-responsive genes. This domain is the VDR element (VDRE). Thus, the binding of the vitamin D-VDR complex to the VDRE results in the transcription of specific mRNAs. In advanced CKD, there are impairments in the binding of vitamin D to VDR as well as in the binding of the vitamin D-VDR complex to the VDRE. Both of these events, in addition to the reduced number of VDR, are responsible for the vitamin D-resistant state of severe kidney dysfunction (Stages 4 and 5).
Bone Disease
The nature and type of bone disease that develops in CKD may vary from one patient to another. Multiple reasons may account for these variations (Table 5). The 2 major types of bone disease that are commonly encountered in patients with CKD are enhanced bone resorption (osteitis fibrosa) and adynamic bone disease. Some patients may have 1 of these types predominantly, whereas others may have a mixed type of bone disease. Mild forms of these derangements in bone metabolism may be observed in the early stages of CKD (Stage 2) and they become more severe as kidney function deteriorates. Osteosclerosis may also occur, and osteoporosis may be encountered.
Bone lesion of excess PTH (high-turnover bone disease). The elevated blood levels of PTH are responsible for the enhanced number and activity of osteoclasts leading to increased bone resorption. As this process increases in severity, marked fibrosis involving the marrow space develops, with the histological picture of osteitis fibrosa becoming evident. The marrow fibrosis is caused by activation of marrow mesenchymal cells, which differentiate into fibroblast-like cells, which form fibrous tissue. In this condition, there is also increased bone formation as evidenced by increased amounts of osteoid. This osteitis fibrosa is a high-turnover bone disease. The manifestations of excess PTH in the bone of uremic patients include increased numbers of osteoclasts and osteoblasts, osteoclastic bone resorption, enlarged haversian lacunae, endosteal fibrosis, and accumulation of woven osteoid and woven bone.

Bone lesion of defective mineralization. Defective mineralization of osteoid leads to rickets in children and osteomalacia in adults. Histologically, osteomalacia can be accurately diagnosed only by the evaluation of undecalcified bone specimens. Osteomalacia is due to a delay in the rate of bone mineralization resulting in accumulation of excess unmineralized osteoid. However, it must be emphasized that the presence of excess osteoid does not necessarily mean osteomalacia. Excess osteoid may be (a) secondary to abnormalities in normal mineralization (osteomalacia); or (b) caused by an increased rate of synthesis of bone collagen, which exceeds normal mineralization. The use of double tetracycline labeling can differentiate between these 2 possibilities and is thus critical for the diagnosis of osteomalacia. The skeleton in osteomalacia is weakened, and patients with this bone disease have skeletal deformities, bone pain, fractures, and musculoskeletal disabilities.
Several mechanisms may underlie the defective mineralization of osteoid and hence the development of osteomalacia in CKD patients. The most important factor in the development of osteomalacia is aluminum overload. Also, relative or absolute deficiency of vitamin D or its active metabolites and/or resistance to their action are factors responsible for the osteomalacia. Vitamin D may affect mineralization through several pathways; it may affect collagen synthesis and maturation, directly stimulate bone mineralization, and/or increase the levels of calcium and phosphorus in the extracellular fluid surrounding the bone. This latter effect is the result of the action of vitamin D on intestinal absorption of these minerals. It is not evident whether a deficiency in one or more of the vitamin D metabolites is critical. For example, few anephric patients with undetectable blood levels of 1,25(OH)2D3 did not show histological evidence of osteomalacia. On the other hand, long-term therapy with 1,25(OH)2D3 improved or healed osteomalacia in many patients with advanced CKD. Osteomalacia may be more frequently encountered in uremic patients with low blood levels of 25(OH)D3.
Second, abnormalities in the formation and maturation of collagen have been found in rats with experimental uremia and in patients with advanced CKD. These derangements result in a defect in collagen cross-linking and may affect bone mineralization. These abnormalities in collagen metabolism are most likely due to vitamin D deficiency. Indeed, treatment with 25(OH)D3 reversed these defects.
Third, inhibition of maturation of amorphous calcium phosphate to its crystalline phase is another defect participating in the genesis of the osteomalacia. The magnesium content of the bones of these patients is increased, and this may interfere with the process of normal mineralization. Magnesium stabilizes the amorphous calcium phosphate and inhibits its transformation into hydroxyapatite. The bone content of pyrophosphate is also increased in these patients, and pyrophosphate may inhibit mineralization.
Fourth, aluminum toxicity may be responsible for a certain type of mineralization defect that is resistant to vitamin D therapy. This type of bone disease has been called low-turnover bone disease or low-turnover osteomalacia. This is mainly seen in dialysis patients who have a large content of aluminum in bone and in whom the aluminum is localized in the mineralization front (ie, the limit between osteoid and calcified tissue). With a decrease in the use of aluminum-containing compounds for the control of hyperphosphatemia, the incidence and prevalence of osteomalacia have been decreasing. Increased burden of iron, alone or in combination with aluminum, can cause osteomalacia in kidney failure patients.
Adynamic bone disease. The exact mechanisms underlying adynamic bone disease (ABD) are not fully elucidated. It is seen in kidney failure patients before and after treatment with peritoneal dialysis or hemodialysis. The prevalence of ABD varies between 15% and 60% in dialysis patients. In 1 study, 30% of bone biopsies from patients with Stage 4 CKD displayed findings consistent with ADB. This entity is characterized by a defect in bone matrix formation and mineralization, increased osteoid thickness, and a decrease in the number of both osteoblast and osteoclast on bone surfaces. There are no excessive amounts of aluminum in the mineralization front. Patients with ABD have lower blood levels of PTH than those with other forms of bone disease. The oversuppression of the parathyroid gland activity with high calcium intake and/or administration of 1,25(OH)2D3 resulting in normal blood levels of PTH may be a factor in the genesis of ABD. ABD is also encountered after parathyroidectomy, in CKD patients with diabetes, and in those with increased aluminum burden; in all these clinical settings, the blood levels of PTH are low. This relationship between the PTH level and ABD is understandable because hypersecretion of PTH in patients with CKD is needed to maintain normal rates of bone formation. It is generally accepted that the blood levels of PTH in the range of 2 to 3 times normal are necessary to maintain normal rates of bone formation and prevent the emergence of ABD.
Patients with ABD have increased rates of overt fractures and microfractures. The latter causes bone pain. Calcium uptake by the adynamic bone is reduced, and therefore patients with ABD may develop hypercalcemia if calcium intake is increased or if dialysate calcium is high.
Osteosclerosis and osteoporosis. Osteosclerosis appears as increased bone density in roentgenographic studies. Histologically, osteosclerosis is most likely due to accumulation of unmineralized trabecular bone with an increase in total bone mass. Because osteosclerosis affects trabecular bone, it is most evident in the vertebrae, pelvis, ribs, clavicles, and metaphyses of long bones, which are made predominantly of cancellous (trabecular) bone. In patients with osteosclerosis, no correlation is found between the bone lesion and any specific pattern of change in serum levels of calcium, phosphorus, or alkaline phosphatase. Certain experimental and clinical evidence suggests that osteosclerosis could be induced by excess PTH. Indeed, patients with primary hyperparathyroidism may display radiographic evidence of osteosclerosis.
Osteoporosis is defined as a decrease in the mass of normally mineralized bone. Immobilization, calcium deficiency per se, and chronic protein depletion may be causes of the osteoporotic component of kidney osteodystrophy. In patients older than 50 years, factors that cause postmenopausal, idiopathic, or senile osteoporosis may contribute to the skeletal abnormalities of CKD.
Role of Acidosis in Bone Disease
Acute acidosis produces a significant loss of the acid-soluble calcium carbonate from bone and is usually associated with negative calcium balance. Rats fed a diet rendering them permanently acidotic were found to have less calcified bone than control animals, despite adequate intake of calcium. Patients with CKD show a persistent positive retention of hydrogen ion which is partially buffered by bone. Indeed, evaluation of the composition of bone in CKD reveals a loss of calcium carbonate. These observations imply that acidosis may contribute to negative calcium balance and the development of skeletal demineralization. However, there is no evidence that the chronic acidosis observed in CKD can produce continued loss of bone minerals once the labile calcium carbonate component of bone is lost. Although there may be a slight improvement in negative calcium balance following treatment of the chronic acidosis with alkali, a positive balance for calcium usually does not occur, and hypocalcemia, bone pain, and radiographic abnormalities are not corrected. Moreover, there is no convincing evidence suggesting that chronic acidosis can cause defective mineralization. It appears that the chronic acidosis of CKD may not play a major role in the pathogenesis of bone disease in adult patients with CKD.
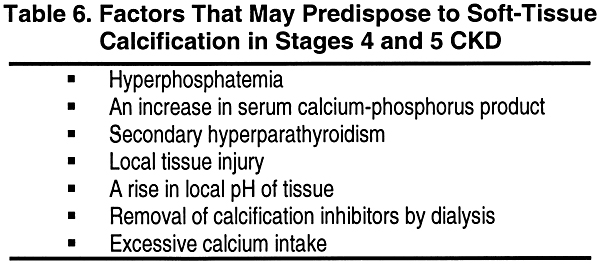
Soft-Tissue Calcification
Various factors present in Stage 4 and 5 of CKD may predispose to soft-tissue calcification (Table 6). An increase in the calcium-phosphorus product in the extracellular fluid is probably the most important pathogenetic factor. The incidence of soft-tissue calcification is high when the calcium-phosphorus product (each in mg/dL) exceeds 70, while soft-tissue calcification is infrequently noted when the calcium-phosphorus product is below 50. These breakpoints not withstanding, and because of the biological variations in range of calcium-phosphorus product over which calcification may occur and because of other contributing factors including age, it is recommended that the product be maintained below 55. Alkalemia, which often occurs after hemodialysis, may persist during the interdialytic period and may predispose to precipitation of calcium salts in soft tissues. An increase in local pH due to loss of CO2 from the exposed part of the eye may bring about the observed conjunctival and corneal calcification. PTH enhances movement of calcium into cells, and the state of secondary hyperparathyroidism may play an important part in the genesis of soft-tissue calcification in kidney failure. Certain factor(s) that may act locally to inhibit calcification and are present in the blood of these patients may possibly be removed during hemodialysis. Local tissue injury may also predispose to calcification when the calcium-phosphorus product is normal or only slightly elevated. The expression of genes coding for certain proteins involved in prevention of calcification has been demonstrated in macrophages and smooth muscle cells of blood vessel walls. One of these proteins is matrix gla protein (MGP). Its deficiency permits medial calcification of blood vessels. Indeed, MGP knockout homozygous mice displayed extensive and severe vascular calcification. It is possible that downregulation of the production of this protein occurs in uremia and participates in the genesis of the vascular calcification seen in patients with kidney failure.
The chemical nature of soft-tissue calcification may vary in different tissues. Thus, the calcification found in nonvisceral tissue (periarticular and vascular calcification) consists of hydroxyapatite, with a molar Ca:Mg:P ratio similar to that of bone. In contrast, the calcification found in visceral organs (skeletal and myocardial muscle) is made of amorphous (CaMg)3(PO4)2 which has a much higher magnesium content. These observations suggest that the mechanisms responsible for the calcification of various tissues in uremic patients may be different.
Soft-tissue calcification constitutes a serious problem in CKD patients. These extraskeletal calcification may be localized in the arteries (vascular calcification), in the eyes (ocular calcification), in the visceral organs (visceral calcification), around the joints (periarticular calcification), and in the skin (cutaneous calcification).
Vascular calcification. Vascular calcification is detected radiographically. The calcification appears as a fine, granular density outlining a portion of the entire artery, giving a radiographic appearance of a pipestem due to deposition of calcium within the media and the internal elastic membrane of the artery. The lumen of the vessel is usually not involved. This medial calcification may first be seen in the dorsalis pedis as a ring or a tube as it descends between the first and second metatarsals. Calcification can also occur in atherosclerotic plaques in the intima of large vessels whose radiographic appearance is that of discrete, irregular densities. It is possible that uremic patients are more prone to this type of calcification because of the presence of hypertension and a propensity to accelerated atherosclerosis.
Arterial calcification is rare in children, uncommon between 15 and 30 years of age, and common in those older than 40. Vascular calcifications are seen in kidney failure patients and in those treated with hemodialysis, and they persist after kidney transplantation. The reported incidence of arterial calcification in dialysis patients has varied from 3% to 83%. In general, the reported incidence of arterial calcification increases with duration of dialysis treatment. In a series of 135 patients published in 1977, the incidence of vascular calcification increased from 27% in those treated for less than 1 year to 83% in patients treated for more than 8 years.
Vascular calcification may involve almost every artery and has been seen in arteries of the forearm, wrist, hands, eyes, feet, abdominal cavity, breasts, pelvis, and brain. The calcification may be very extensive, rendering the artery so rigid that the pulse is not palpable and the Korotkoff sounds may be difficult to hear during the measurement of the blood pressure. Such calcification may also present difficulties during surgery for the creation of arteriovenous shunts or fistulas for maintenance hemodialysis or during renal transplantation.
Arterial calcification shows little tendency to regress; in some patients, improvement or disappearance of arterial calcification occurs within months to years after subtotal parathyroidectomy or renal transplantation.
Ocular calcification. Ocular calcification are the most common types of soft-tissue calcification seen in Stage 4 and 5 CKD. Calcium deposition in the eye may produce visible inflammation and local irritation, resulting in the red eye of uremia. This is a transient phenomenon and may last only a few days. Recurrence of the red eye phenomenon is not infrequent, and it becomes apparent each time a new calcium deposition occurs in the conjunctiva. More commonly, conjunctival calcium deposits are asymptomatic and are seen as white plaques or as small punctate deposits on the lateral or medial segment of the bulbar conjunctiva. Also, calcium deposits may occur within the cornea at the lateral or medial segments of the limbus, the so-called band keratopathy. Slit-lamp examination permits easier recognition of these lesions. The loss of CO2 through the conjunctival surface into the air increases the local pH of the ocular tissue, and this rise in pH predisposes to calcium deposition.
Visceral calcification. Deposits of calcium may be found in the lungs, stomach, myocardium, skeletal muscles, and kidney. These calcification are usually not evident radiographically, but can be detected by 99mTc-pyrophosphate scan.
Visceral calcification may cause serious clinical complications. Congestive heart failure, cardiac arrhythmias, and heart block may occur in patients with calcium deposition in the myocardium or in and around the conduction system of the heart or the mitral annulus. Calcification of cardiac valves are not infrequent. Abnormal pulmonary function may be noted in patients with pulmonary calcification. Such patients may have reduced vital capacity and reduced carbon monoxide diffusion. Improvement in pulmonary function has been noted after subtotal parathyroidectomy in these patients. Extensive pulmonary calcification may lead to severe pulmonary fibrosis, pulmonary hypertension, and right ventricular hypertrophy. Calcification of the heart and lung constitute a major risk factor for increased morbidity and mortality in dialysis patients.
Increased oxalate burden may occur in Stage 5 CKD patients, especially if they receive large amounts of ascorbic acid. This may be associated with marked deposition of calcium oxalate in soft tissues. Such deposition in the myocardium, or mitral and aortic valves, can cause cardiomyopathy and congestive heart failure, eventually leading to death. Since vitamin C is metabolized to oxalic acid, it is recommended that vitamin C intake in Stage 5 CKD patients be limited to the daily recommended dose.
Periarticular calcification. Periarticular calcification, with or without symptoms, may develop in patients with Stage 5 CKD. The incidence of periarticular calcification varies widely among dialysis patients. These calcification were absent in 1 report but were encountered in up to 52% of the patients in other series of dialysis patients. The incidence of periarticular calcification may increase with the duration of dialysis. In a study of 135 patients, the incidence of these calcification increased from 9% to 42% from the first to the eighth year of dialysis. With better control of serum levels of phosphorus, this type of calcification is not encountered frequently.
Periarticular calcification may be detected because of the pain induced by the deposition of calcium or may be noted by routine X-ray examination. Most frequently, the calcification appear as small discrete radiodensities around the shoulders, wrists, phalangeal joints, hips, or ankles. Tendosynovitis or tendonitis with abrupt pain may develop, presumably caused by deposition of microcrystals of hydroxyapatite. The synovial fluid of the involved joints is clear with normal viscosity and number of cells. This acute periarticular illness is called calcific periarthritis.
Occasionally, large tumoral masses consisting of encapsulated chalky fluid or pastelike material develop adjacent to joints of dialysis patients. The lesions are usually painless, but they may restrict movement of the joint by virtue of their size. The intake of food with high phosphorus content may enhance the development of tumoral calcification. These lesions often regress with the control of serum phosphorus levels by phosphate-binding antacids or following subtotal parathyroidectomy.
Cutaneous calcification. These lesions may appear as small macules or papules composed of firm calcium deposits which are best detected by the chemical analysis of small skin biopsy specimens. Calcium content of skin is increased in most uremic patients and such increments are more commonly seen in patients with severe secondary hyperparathyroidism. Subtotal parathyroidectomy is followed by a decrease in the calcium content of skin, underscoring the role of secondary hyperparathyroidism in the genesis of the cutaneous calcification. Children exhibit soft-tissue calcification far less frequently than adults, and the calcium content of skin is significantly lower than that observed in adults.
Skin ulcerations and tissue necrosis. A syndrome characterized by the development of progressive ischemic skin ulcerations involving the fingers, toes, thighs, legs, and ankles has been observed in a small number of patients with advanced kidney failure. This syndrome occurs in patients after successful kidney transplantation, in those treated with hemodialysis, and less frequently in patients with Stage 5 CKD who are not on dialysis. It appears that this entity is less common among patients treated with continuous ambulatory peritoneal dialysis.
The patients almost always have vascular calcification involving the media of the arteries, and they usually exhibit X-ray evidence of subperiosteal bone resorption. Serum calcium is usually normal and occasionally elevated. A period of hyperphosphatemia has been present for some time before the appearance of the syndrome. The lesions may be preceded or accompanied by severe pain. Before the appearance of ulcerations or tissue necrosis, tender, slightly erythematous, subcutaneous nodules may develop, or there may be blotchy, bluish discoloration. Raynaud’s phenomenon may also precede the lesions of the fingers or toes. The ulcers may develop slowly over several months, or may appear and progress rapidly over a few weeks. Infection may supervene, leading to sepsis and death. The original reports of this entity termed it calciphylaxis because of an apparent similarity to the calciphylaxis described by Seyle in 1962. Others have argued that the name should be changed to calcific uremic arteriopathy.
This syndrome could be life-threatening and requires aggressive therapeutic attention. The lesions do not respond to treatment with local measures but have healed following subtotal parathyroidectomy in most patients. However, in some patients, the lesions did not heal after parathyroidectomy, and in others, the lesions seem to be aggravated.
Although disturbances in mineral metabolism, secondary hyperparathyroidism, and vascular calcification appear to play an important role in the genesis of this entity, other factors may also contribute to its emergence and progression. Acquired protein C deficiency has been reported in patients with CKD, and such a derangement may lead to a hypercoagulability state and consequently to vascular occlusion and tissue necrosis. Therefore, it is important that the blood levels and the activity of protein C be measured in patients with calciphylaxis. It is interesting that obesity, especially in white women, predisposes to calciphylaxis, and the relative risk for calciphylaxis rises with weight increase. Local trauma may be a contributory factor as to site where the lesion may appear; indeed, in some patients, the necrotic lesions began in areas where insulin, heparin, or iron dextran were injected. Warfarin prescription is a possible risk factor as well.
Disturbances in mineral and bone metabolism are common in patients with CKD. The processes causing disordered mineral metabolism and bone disease have their onset in the early stages of CKD, continue throughout the course of progressive loss of kidney function, and may be influenced beneficially or adversely by various therapeutic approaches used. The pathogenesis of abnormalities in bone mineral metabolism and disease in CKD are shown in Fig 1.
A large body of evidence has accumulated indicating that the derangements in mineral and bone metabolism in CKD are associated with increased morbidity and mortality. These patients have bone pain, increased incidence of fractures, bone deformity, myopathy, muscle pain, and ruptures of tendons, and children with chronic kidney failure suffer from retarded growth.
The long-term effects of soft tissue calcifications have become an area of growing concern for CKD patients and those who treat them. Calcification of the lung leads to impaired pulmonary function, pulmonary fibrosis, pulmonary hypertension, right ventricular hypertrophy, and right-side congestive heart failure. Calcification of the myocardium, coronary arteries, and cardiac valves result in congestive heart failure, cardiac arrhythmias, ischemic heart disease, and death. Vascular calcification leads to ischemic lesions, soft-tissue necrosis, and difficulties for kidney transplantation.
Hyperphosphatemia also appears to be associated with increased mortality, and elevated blood levels of PTH exert significant adverse effects on the function of almost every organ. Thus, prevention of the disturbances in mineral and bone metabolism and their management early in the course of chronic kidney disease are extremely important in improving patients’ quality of life and longevity.
Although much remains to be learned about these conditions, the recommendations made in these guidelines are intended to aid clinicians in developing an integrated approach to their diagnosis and management of this complicated area, based on the best available evidence. It is clear that the kidney community has many opportunities to develop strategic alliances in order to add to the existing body of knowledge. Ongoing research in this exciting area will lead to improvements in care and, thus, to updating of guidelines when such information is available.
Throughout the guidelines, the stages of CKD are defined according to the K/DOQI Clinical Practice Guidelines for Chronic Kidney Disease: Evaluation, Classification, and Stratification (Table 2)